Cutevariant support Variant Call Format (VCF) Version 4.2. It is strongly advised to annotate your VCF file in a second pass using annotation tool like VEP or SnpEff. Annovar is yet not supported.
Quick example#
Create a new project#
We will use the same protocol as SnpEff which is described on this page.
As mentioned, the aim is to find a pathogenic mutation in a family of 17 individuals where 3 (in red) are affected by an homozygous inheritance disease.
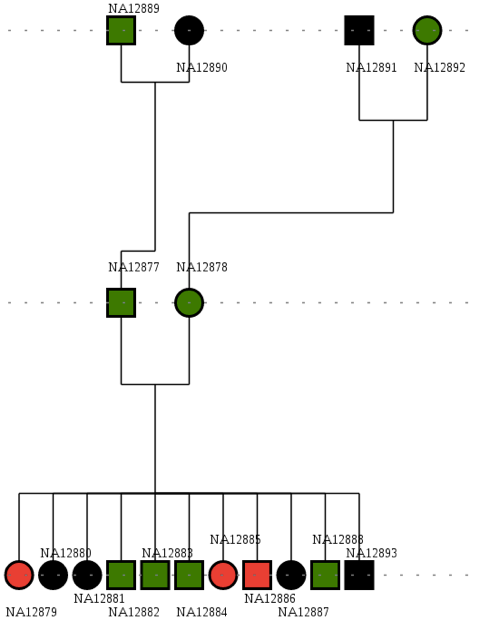
Download dataset#
We provide here the VCF file annotated with SnpEff with the corresponding ped file.
Create a new project#
After cutevariant is launched, click on new project on the upper toolbar and follow the instructions from the wizard. You will import the VCF file with the ped file describing affected and non affected samples. Depending of the size of the file, it can take several minutes to be imported into sqlite database. But keep in mind, this step is performed only once. After the database creation, you can open the sqlite file directly with the open project button.
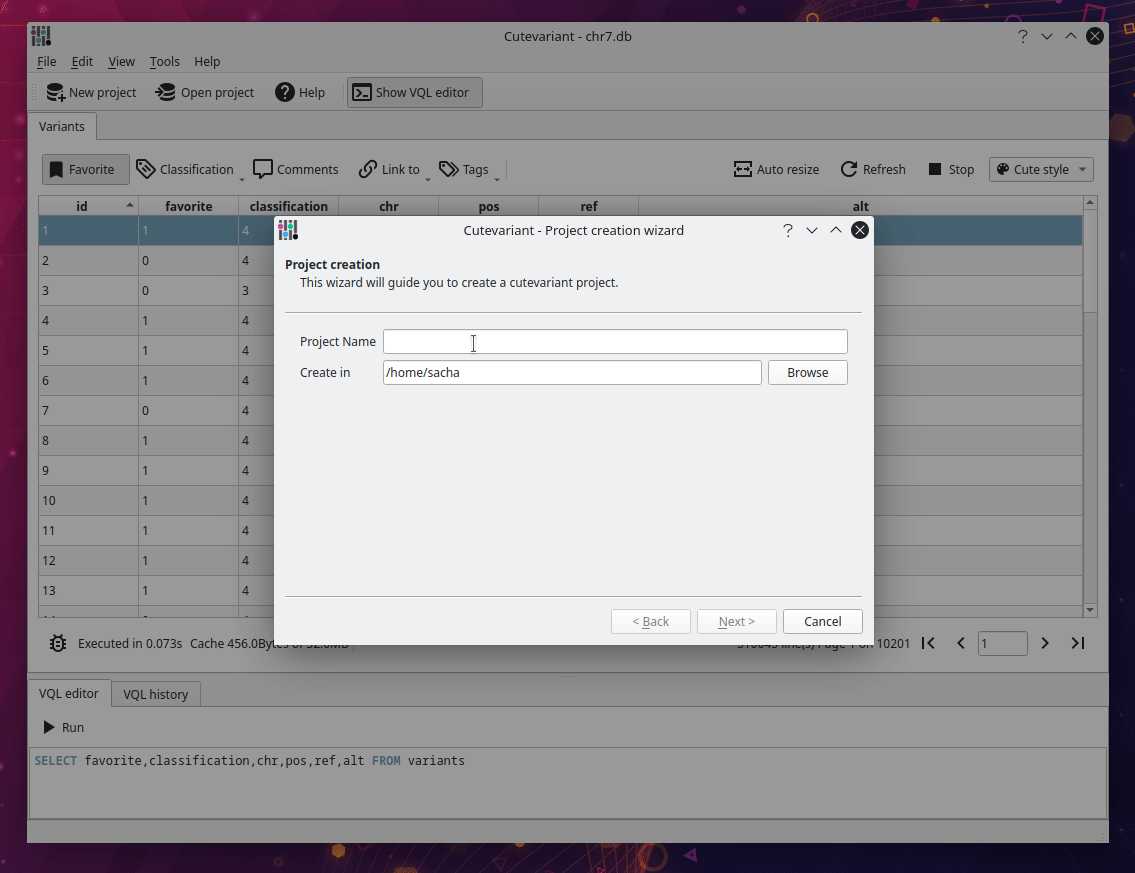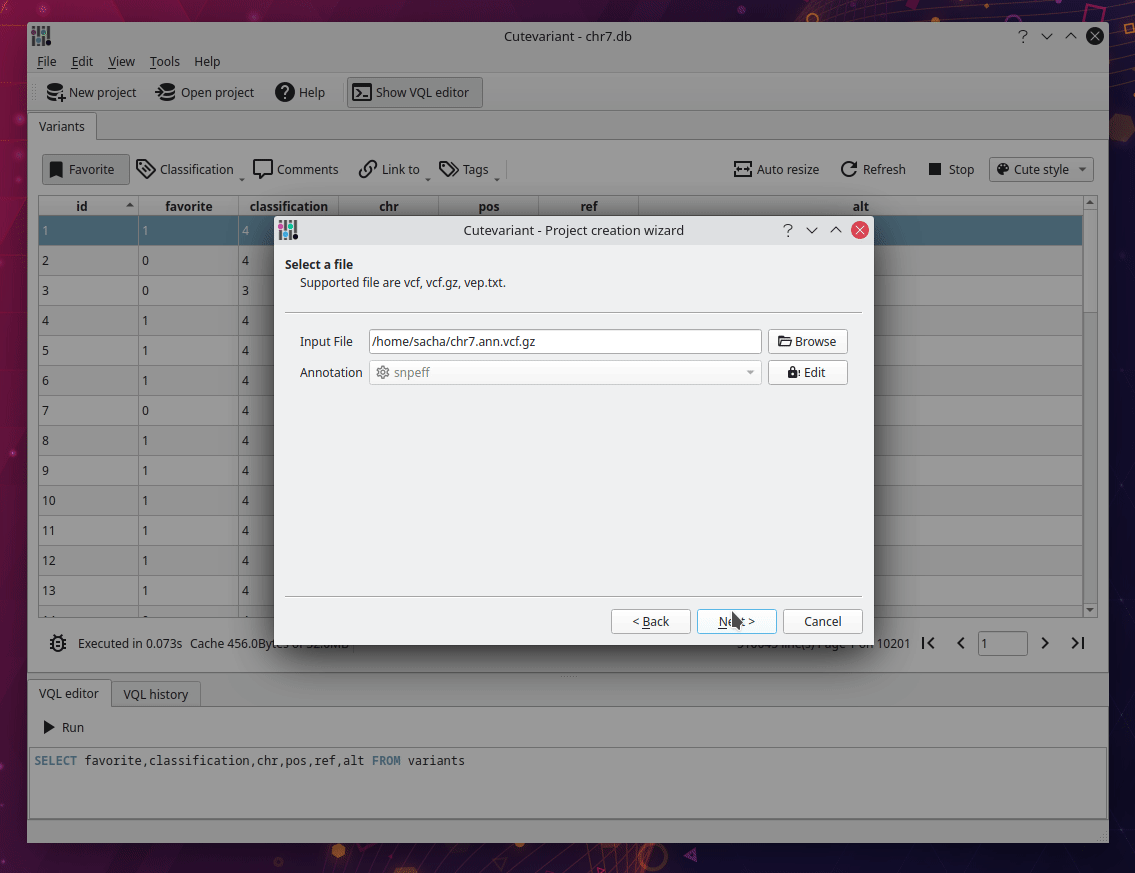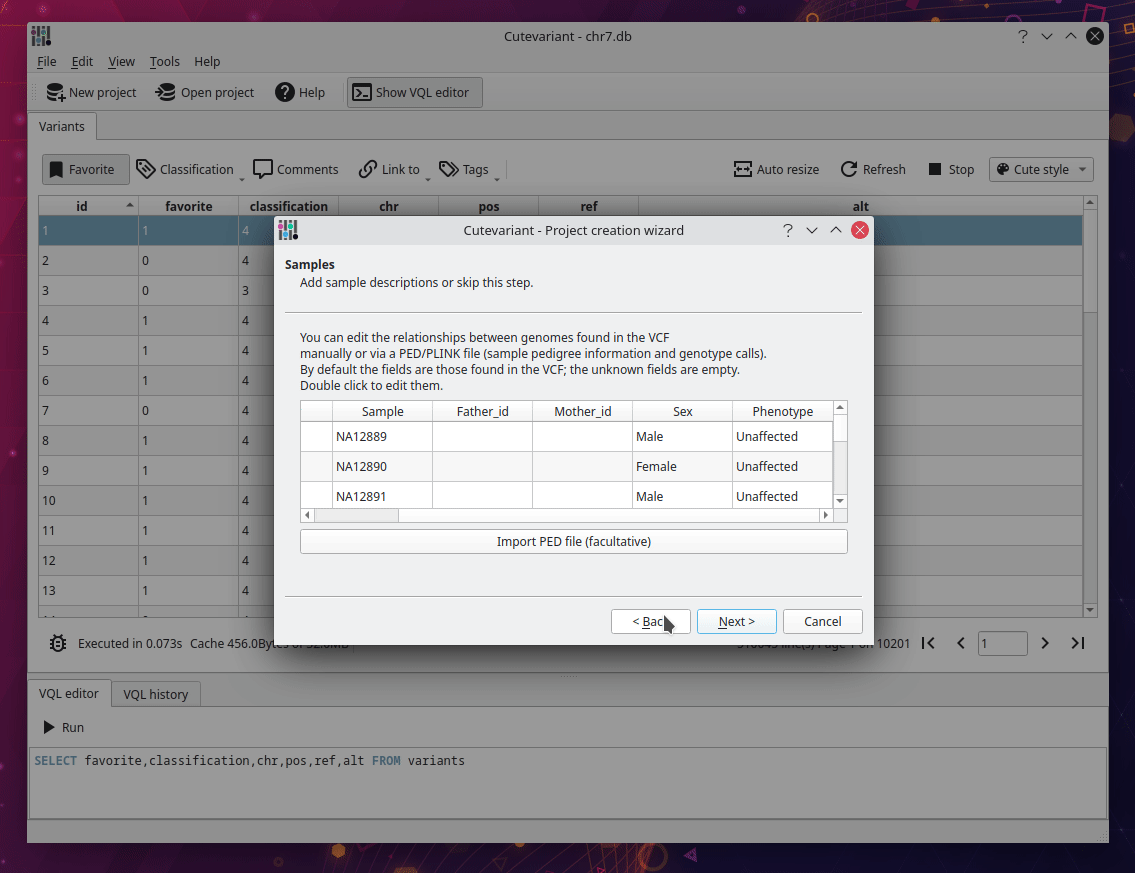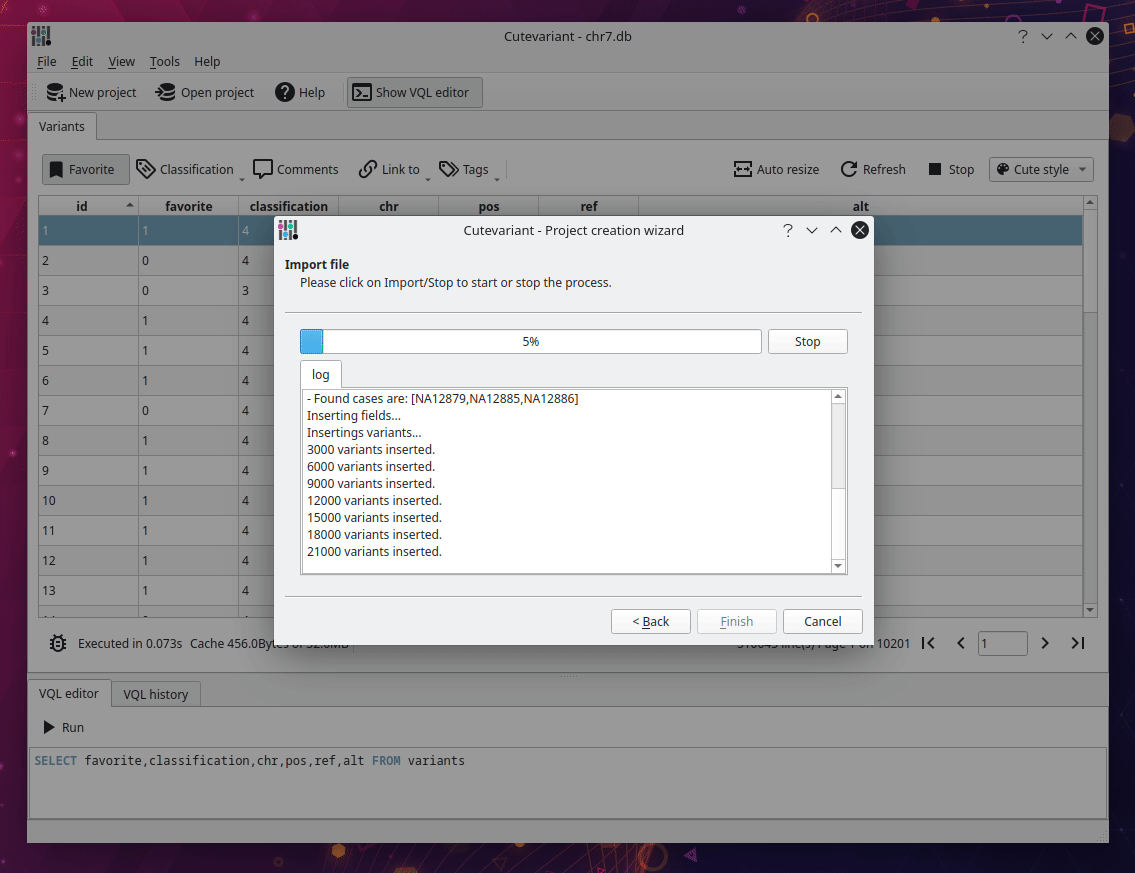
Select fields to display#
You should now see the following view displaying a table of all variants loaded from the VCF file. Different plugins are available around to control the view and filtering in different way. They are available from view menu. But you are encouraged to use the VQL which is much faster. For instance, write the following VQL query to select interesting fields:
SELECT chr, pos, ref, alt, ann.gene, ann.impact FROM variants
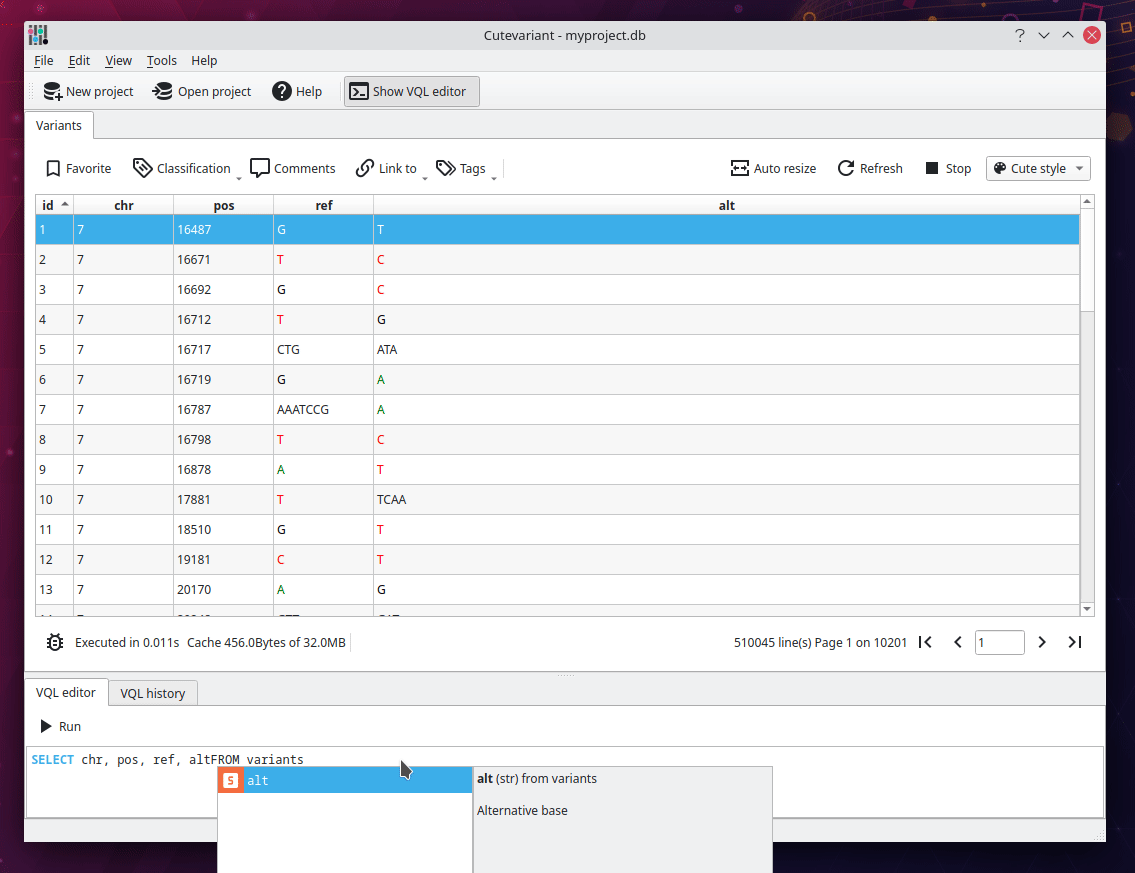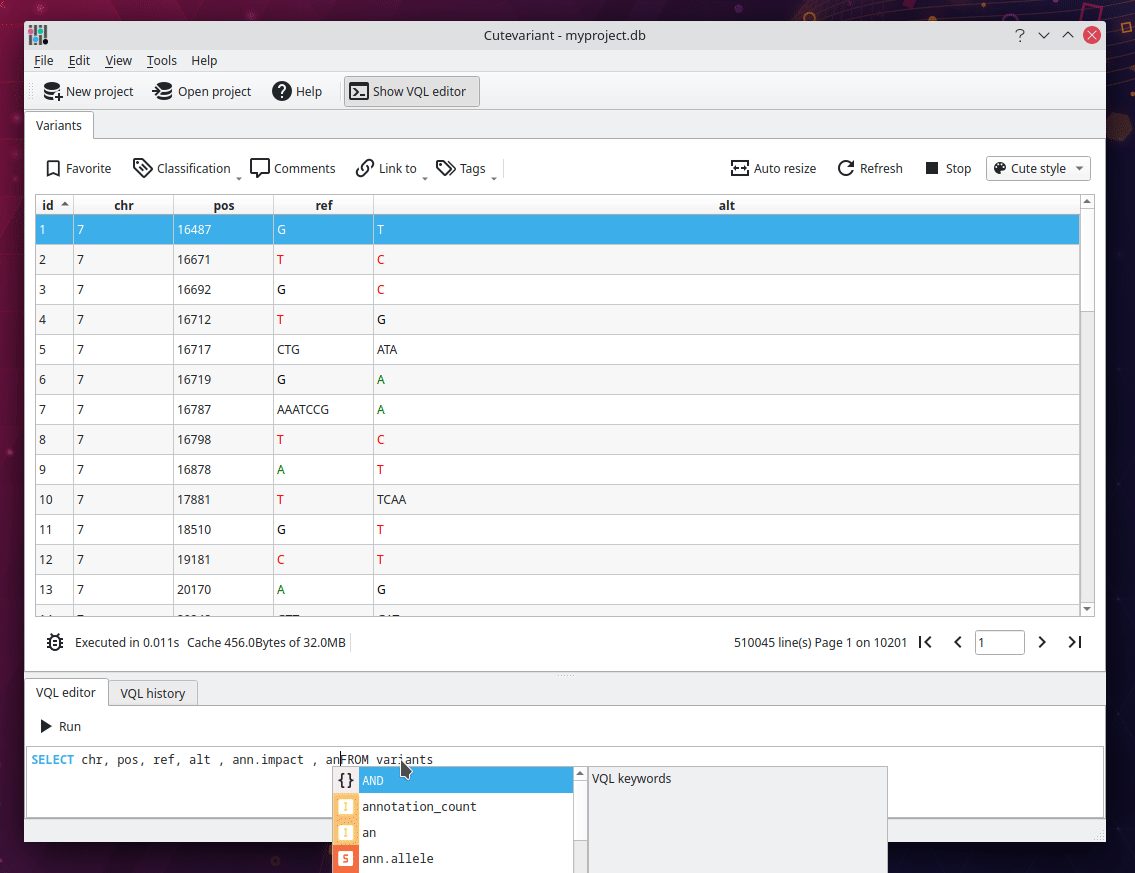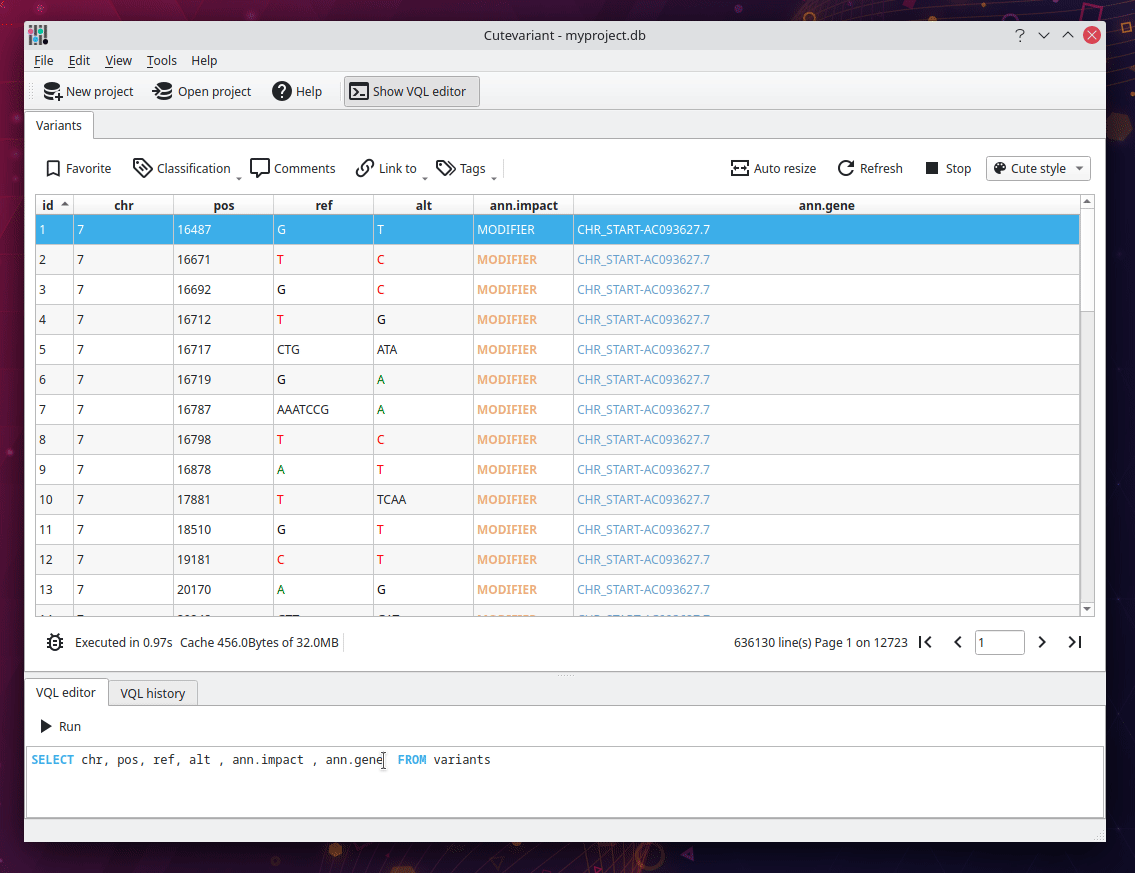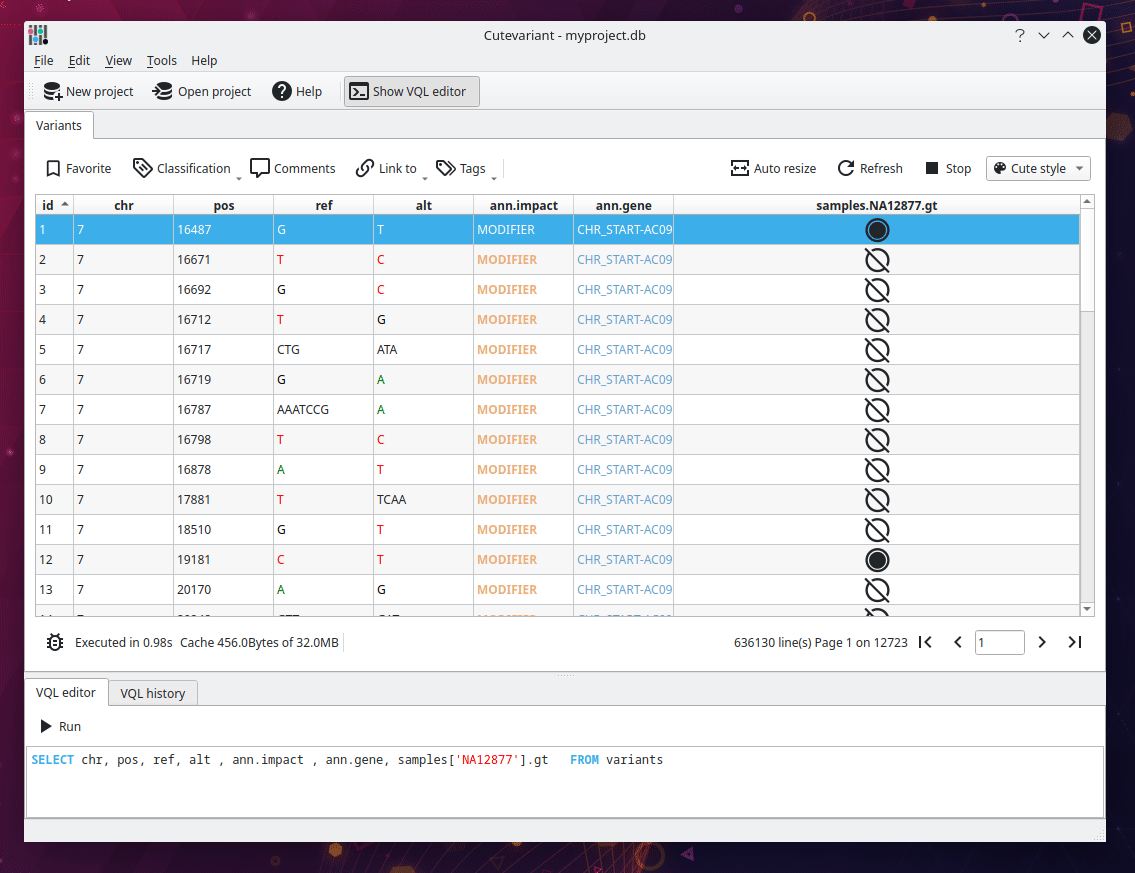
Filter variant with VQL#
From the original SnpEff documentation, we have to find all variant with HIGH impact where three affected samples are homozygous mutated and the unaffected are not. Cutevariant generates special fields for each variant, telling the count of sample with a specific genotype. For instance, the field "control_count_hom" return the number of affected samples that are homozygous. In brief, the following query ask "Give me all variants with HIGH impact which are muted homozygous in 3 affected samples (case) and wild homozygous in 0 unaffected samples (control)".
SELECT favorite,comment,chr,ref,alt,ann.consequence,ann.impact,ann.gene FROM variants
WHERE case_count_hom = 3.0 AND control_count_hom = 0.0 AND impact = 'HIGH'
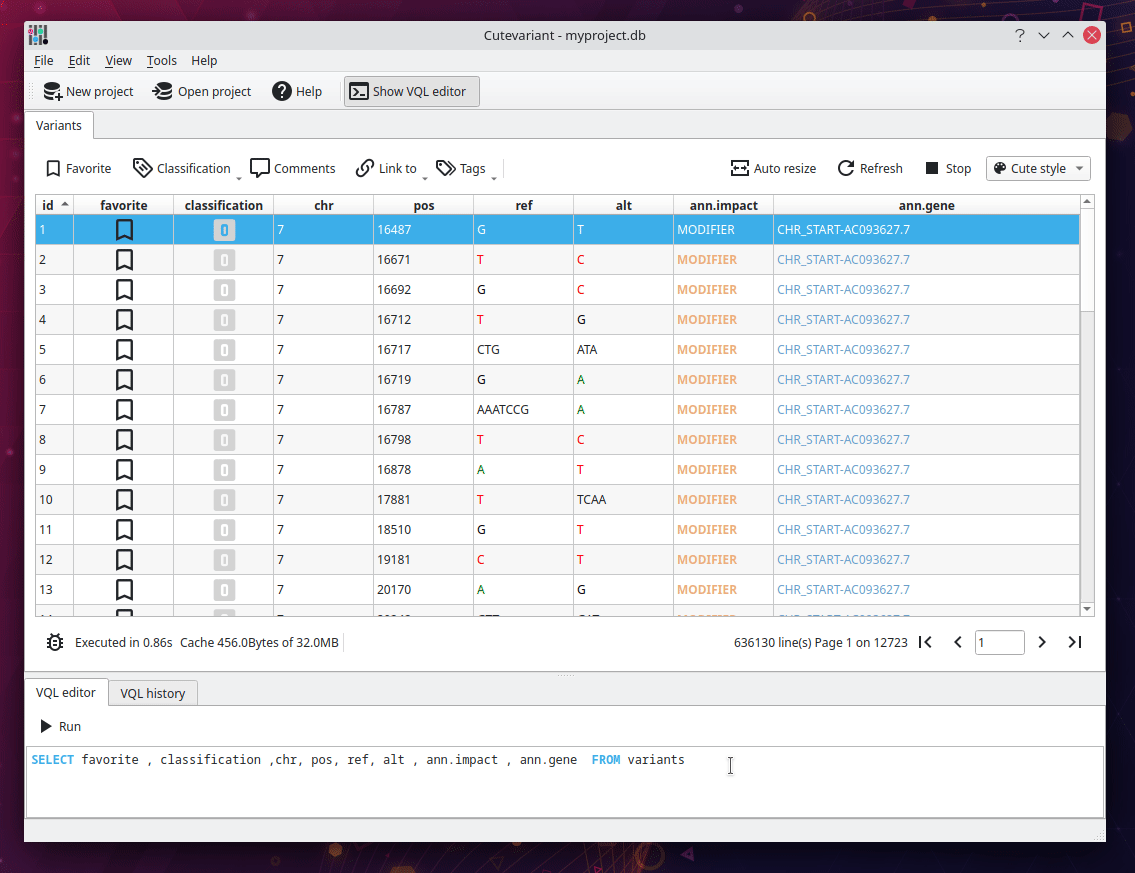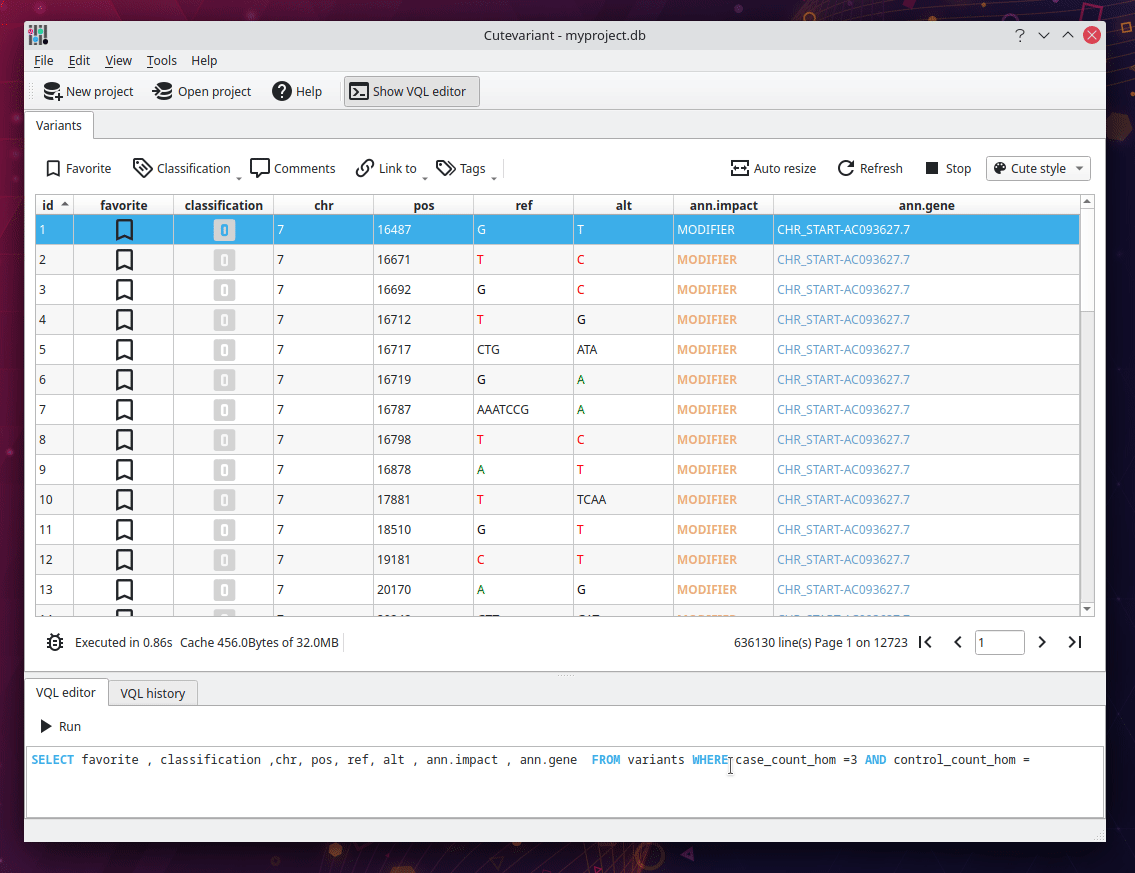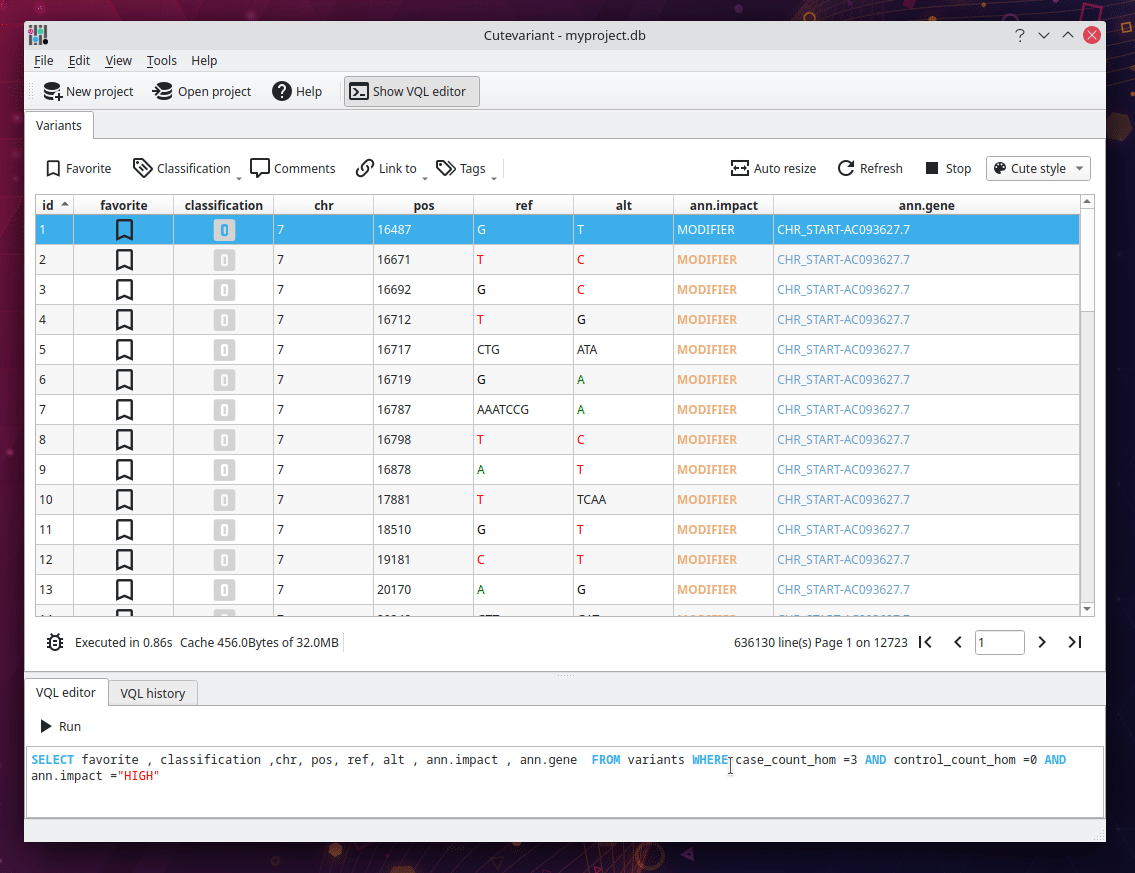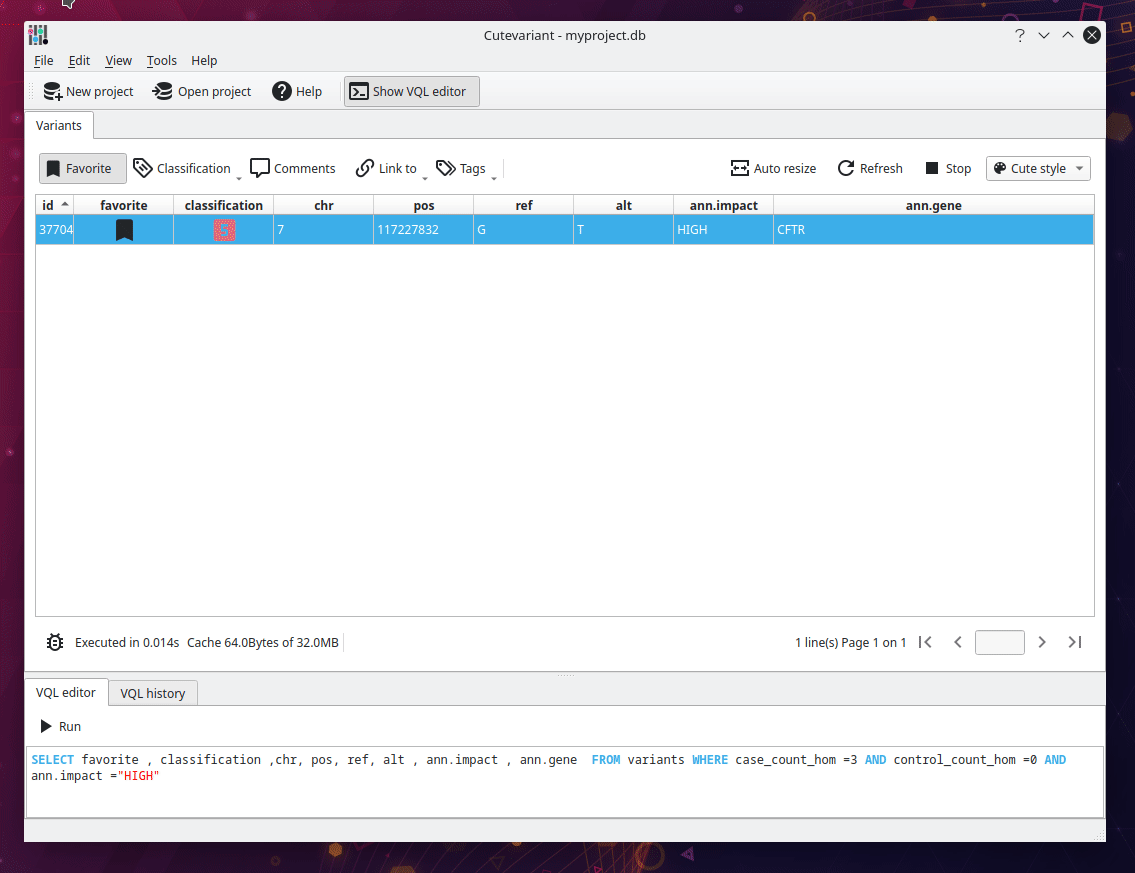
Congratulation, you succeeded to identify the variant in no time !